
Kia ora koutou,
Many have become familiar with a new term over the recent COVID-19 outbreak in Aotearoa New Zealand – ‘whole genome sequencing’. Genome sequencing creates a ‘genetic fingerprint’ of the COVID-19 virus that has infected an individual. It can tell us whether the virus that has infected two different people has a closely related or different genetic sequence, which is hugely valuable to support contact tracing, cluster analysis and understanding viral evolution. Genome sequencing has proven to be a critical tool in the management of this second outbreak in Auckland.
During the first wave of COVID-19 in Aotearoa, people were returning home from all over the world and brought with them different variants of the virus. Local researchers sequenced the COVID-19 genomes of 649 of these cases and identified nearly 300 different introductions from different parts of the world. With so many seed cases, this outbreak had the potential to escalate very quickly all over Aotearoa.
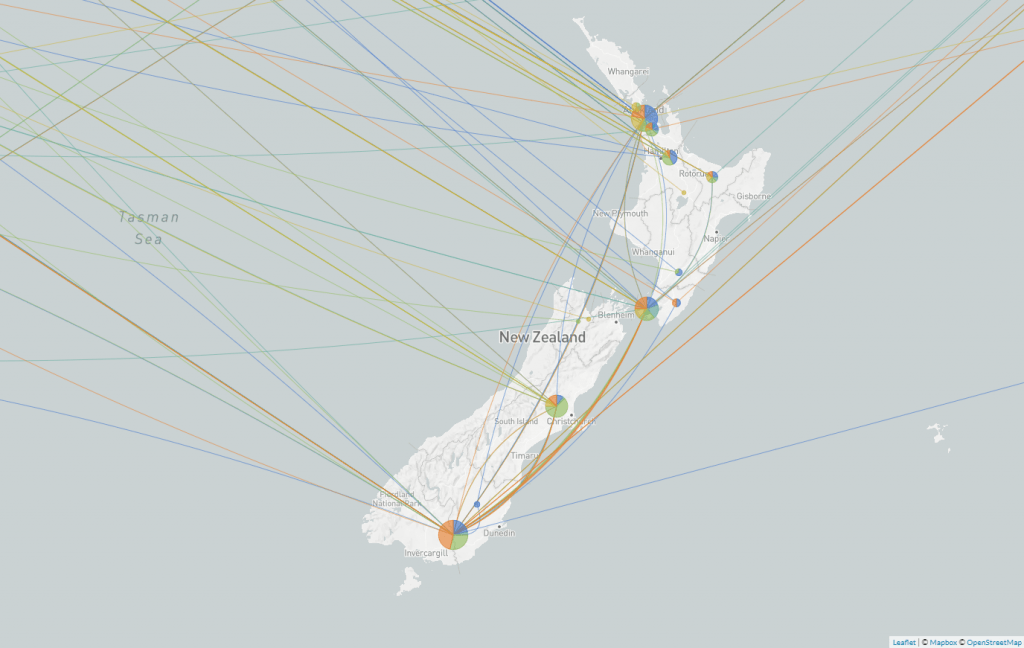
Screengrab of a map from Nextstrain showing the different variants of SARS-CoV-2 in Aotearoa New Zealand.
During the second outbreak, our use of genome sequencing has become more integral, informing management of the cluster in real time. Scientists at ESR have been working hard to rapidly sequence the genomes of new cases and use this information to link cases to and within the cluster.
There were several instances where we were reassured that seemingly unconnected clusters were actually closely linked. There were other times when the specific mutation in a sequence significantly narrowed the search for the contact tracers. Without that information, we would have been in the dark about whether new cases lacking a known link to the cluster were due to another source (e.g. a managed isolation facility) or were in fact linked to the community outbreak. Just imagine how much harder it would have been to manage the second outbreak without this tool, with cases popping up right across Auckland.
With the genome sequencing in place, we knew that the broad geographical spread simply reflected the way people live in a city – crossing suburbs widely and connecting across town rather than staying in their postcodes. It gave us rich information about the sprawling nature of our biggest cluster(s) and allowed us to understand why more localised lockdowns, such as those used in Melbourne, were unsuccessful.
The next challenge is to continue to improve our processes so that an analysis of the viral genome is embedded in our COVID-19 response for every case and at pace. This includes rapid transport of samples to a sequencing facility, even in scenarios when planes aren’t flying often out of the region.
Observations and recommendations stemming from these powerful new tools – including what we have achieved and how we can keep improving – are covered in a new rapid review by Professor Michael Bunce, Chief Scientist of the EPA.
You can read the rapid review on the role of genomics here (PDF, 5MB)
A huge congratulations to the scientists at ESR who led this excellent work and helped us get on top of the Auckland outbreak. Ka rawe!
Noho ora mai,

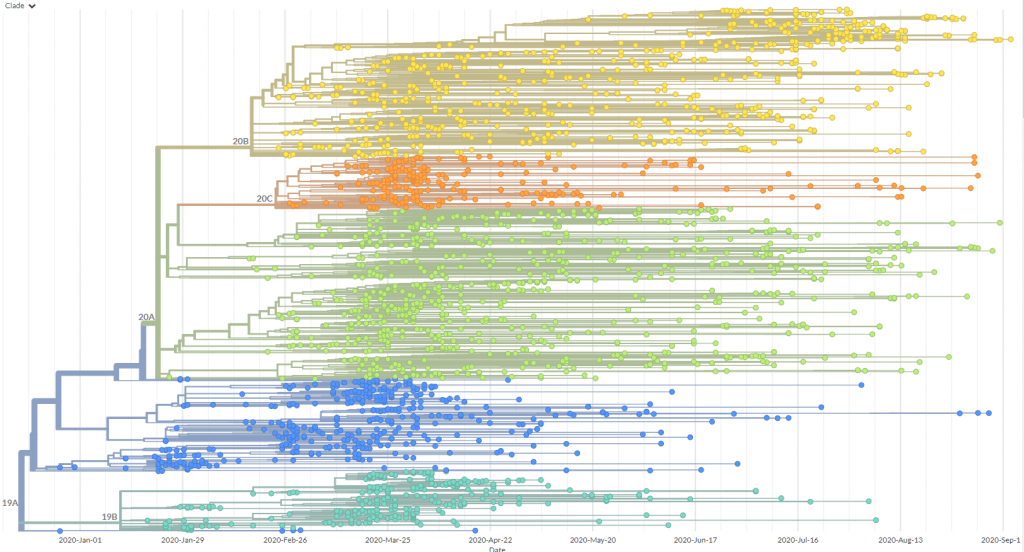
Screengrab from Nextstrain of a phylogenetic tree visualising SARS-CoV-2 variants from Oceania.